
Abbreviations: cfDNA, cell free DNA; ddPCR, droplet digital polymerase chain reaction; NGS, next-generation sequencing; PD, progressive disease.
Improved Turnaround Times for Biopsy Results Matters More Than Type of Biopsy
By Dr. Edgardo S. Santos Castillero, MD, FACP
Posted: December 11, 2019

Castillero
Regarding “Liquid Biopsy for Assessing Response or Progression in Advanced NSCLC” by Dr. Geoffrey R. Oxnard published in the October issue of the IASLC Lung Cancer News, I would like to comment on the issue of turnaround time (TAT), which is a topic that is extremely critical in the treatment of patients with cancer.
Significant improvement has been observed regarding TAT since the Guardant360 became commercially available in 2014. This test uses next-generation sequencing (NGS) to determine the genomic alterations in blood after cell-free DNA (cf-DNA) from cancer cells is captured. Initially, the test consisted of 58 genes, with a TAT of approximately 10-12 calendar days. The test was increased to 73 genes, but its TAT has been reduced to only 7 calendar days as demonstrated in the prospective NILE study.1 The first 10 patients had a median cf-DNA TAT of 14 days (range 11-30 days) versus the last 10 patients, who had a median TAT of 7 days (range 5-9 days). As a pioneer in this field, the Guardant Health test has been challenged by other competitors, such as Biodesix’s Genestrat, which uses digital droplet polymerase chain reaction (ddPCR) technology and has an average TAT of 3 days. This test analyzes six actionable genomics abnormalities (EGFR, ALK, ROS1, RET, BRAF, and KRAS) in lung cancer. Inivata also uses NGS with its InvisionFirst-Lung test to analyze 36 genes relevant to patients with advanced NSCLC, with a TAT of 7 calendar days or fewer from blood draw. As a clinician, this is amazing. We can diagnose driver mutations, resistant mutations, and, perhaps in the future, monitor our patients in a seamless manner. Only rarely do we have to rush to change therapy for a patient who has been well monitored. Hence, to wait just 3-7 calendar days to obtain a “genomic snapshot” of our patients is probably as good as it gets. The TAT for NGS in tumor tissue has also significantly improved, with some vendors in the private sector reporting NGS in 5 calendar days. In my opinion, to wait more than 14 calendar days for an NGS report, regardless of whether derived from blood or tissue specimen, is unacceptable. ✦
Reference:
1. Leighl NB, Page RD, Raymond VM, et al. Clinical Utility of Comprehensive Cell-free DNA Analysis to Identify Genomic Biomarkers in Patients with Newly Diagnosed Metastatic Non-Small Cell Lung Cancer. Clin Cancer Res. 2019;25(15):4691-4700.
The Application of Liquid Biopsy in Lung Cancer: The View from China
By Fei Zhou, MD, and Caicun Zhou, MD, PhD
Posted: December 11, 2019


The application of liquid biopsy in lung cancer is increasing in China. For patients with insufficient tumor tissue, liquid biopsy could provide tumor cell-derived genomic landscape for precision therapy. Due to its minimally invasive nature, liquid biopsy can also be repeated serially for longitudinally monitoring treatment response, detecting the emergence of drug resistance, and tracking tumor evolution.1 Here, inspired by the October IASLC Lung Cancer News article “Liquid Biopsy for Assessing Response or Progression in Advanced NSCLC” by Dr. Geoffrey R. Oxnard, we briefly discuss how liquid biopsy has been used in China, as well as its challenges and drawbacks.
The successful development of targeted therapy for advanced NSCLC is based on molecular classification, including EGFR sensitizing mutations, ALK/ ROS1/RET rearrangements, and BRAFV600E mutations. Genomic profiles are recommended to be evaluated in treatment-naive patients with advanced NSCLC in China, especially in those with non-squamous NSCLC. For patients with insufficient tumor tissue, circulating-free DNA (cfDNA) or circulating-tumor DNA (ctDNA) is strongly recommended for EGFR mutation detection according to guidelines by the Chinese Society of Clinical Oncology (CSCO). Super-ARMS has been approved by National Medical Products Administration (NMPA) for EGFR mutation detection using ctDNA. Other validated PCR-based assays, including cobas and digital droplet PCR (ddPCR) are also acceptable. CSCO guidelines also moderately recommend validated next-generation sequencing (NGS) multiplex panels for initial molecular diagnosis using ctDNA for patients without obtainable tumor tissue specimens.
For patients for whom first- or second-generation EGFR-TKI treatment fails, T790M secondary mutation is the predominant acquired resistance mechanism,2 which can be effectively inhibited by the third-generation EGFR-TKI osimertinib.3 ctDNA has been advocated as a feasible source to identify T790M mutations, as a complement to routine tissue-based genotyping according to the guidelines. ctDNA is also clinical available to comprehensively understand the acquired resistance mechanisms of osimertinib to further guide subsequent personalized therapy.4,5 Furthermore, dynamic monitoring of ctDNA or T790M mutations in plasma is being used to predict efficacy and clinical outcomes of patients treated with EGFRTKIs, allowing longitudinal monitoring of patients during treatment.6,7 However, serial ctDNA for assessing response or progression has not been used routinely in China, just serving as a complement to imaging and clinical evaluation or for scientific purposes.
Despite considerable promising advances that liquid biopsy has made, challenges remain. For EGFR mutation detection using ctDNA, although high specificity and concordance with tumor tissue have been achieved with super-ARMS, sensitivity varies from 76%-82%.8,9 Th erefore, negative results should be interpreted with caution, and a more sensitive method (such as ddPCR or NGS approach) or using DNA from tissue biopsy should be performed to rule out false negatives. On the other hand, NGS-based approaches could provide more comprehensive molecular profiles of tumors rather than PCR-based approaches; however, no NGS approach in oncology has been approved by NMPA currently. Furthermore, NGS is still costly and time-consuming, requiring approximately 2 weeks for results turnaround. Finally, we agreed with Dr. Oxnard that some other challenges, including how best to quantify mutations levels and define meaningful changes of ctDNA for monitoring treatment, must still be addressed to ensure reliable treatment decisions in the clinical setting. ✦
About the Authors: Dr. Fei Zhou is in the Department of Medical Oncology, Shanghai Pulmonary Hospital, Thoracic Cancer Institute, Tongji University School of Medicine, Shanghai, China. Dr. Caicun Zhou is director and professor of medical oncology, Shanghai Pulmonary Hospital, Thoracic Cancer Institute, Tongji University School of Medicine, Shanghai, China.
References.
1. Siravegna G, Marsoni S, Siena S, et al. Integrating liquid biopsies into the management of cancer. Nat Rev Clin Onc. 2017;14:531-548.
2. Ohashi K, Maruvka YE, Michor F, et al. Epidermal growth factor receptor tyrosine kinase inhibitor-resistant disease. J Clin Oncol. 2013;31:1070-1080.
3. Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N Engl J Med. 2017;376:629-640.
4. Chen K, Zhou F, Shen W, et al. Novel Mutations on EGFR Leu792 Potentially Correlate to Acquired Resistance to Osimertinib in Advanced NSCLC. J Thor Onc. 2017;12:e65-e68.
5. Yang Z, Yang N, Ou Q, et al. Investigating Novel Resistance Mechanisms to Third- Generation EGFR Tyrosine Kinase Inhibitor Osimertinib in Non-Small Cell Lung Cancer Patients. Clin Canc Res. 2018;24:3097-3107.
6. Wang Z, Cheng Y, An T, et al. Detection of EGFR mutations in plasma circulating tumour DNA as a selection criterion for first-line gefitinib treatment in patients with advanced lung adenocarcinoma (BENEFIT): a phase 2, singlearm, multicentre clinical trial. Lancet Resp. 2018;6:681-690.
7. Zhou Q, Yang JJ, Chen ZH, et al. Serial cfDNA assessment of response and resistance to EGFRTKI for patients with EGFR-L858R mutant lung cancer from a prospective clinical trial. J Hem Onc. 2016;9:86.
8. Feng WN, Gu WQ, Zhao N, et al. Comparison of the SuperARMS and Droplet Digital PCR for Detecting EGFR Mutation in ctDNA From NSCLC Patients. Trans Onc. 2018;11:542-545.
9. Li Y, Xu H, Su S, et al. Clinical validation of a highly sensitive assay to detect EGFR mutations in plasma cell-free DNA from patients with advanced lung adenocarcinoma. PloS One. 2017;12:e0183331.
Liquid Biopsy for Assessing Response or Progression in Advanced NSCLC
By Geoffrey R. Oxnard, MD
Posted: November 12, 2019

Genomic analysis of plasma cell free DNA (cfDNA) has been adopted widely in academic cancer centers (but not necessarily in community settings) for convenient genotyping of advanced NSCLC. Indeed, the rapid uptake of this novel diagnostic as part of the standard of care reflects the compelling nature of such a convenient molecular assay. Intuitively, many are now asking how these blood tests can be used for guiding cancer care once a treatment decision has been made.
For example, it would be valuable if we could use these blood tests to monitor treatment outcomes in the same way that serum tumor markers (e.g., CEA, CA19- 9, and CA125) are used for some cancer types. In contrast to proteins found in serum, plasma cfDNA has a short half-life; therefore, levels can change quickly. We and others have shown that mutation levels in plasma cfDNA can decrease dramatically during treatment and can increase in advance of disease progression.1,2 These dynamics can be detected either with focused assays like droplet digital polymerase chain reaction (ddPCR) or with multigene assays like next-generation sequencing (NGS; Fig. 1).
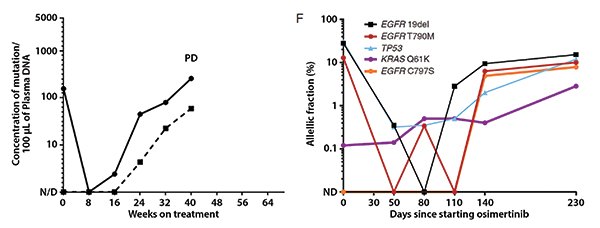
Abbreviations: cfDNA, cell free DNA; ddPCR, droplet digital polymerase chain reaction; NGS, next-generation sequencing; PD, progressive disease.
Despite these observations, some major challenges remain before treatment monitoring using plasma cfDNA can be adopted clinically.
How Best to Quantify Mutation Levels
Early assays measured the absolute concentration of mutant DNA in plasma, quantified as copies/mL.1 With the emergence of NGS assays, it is now more common to quantify the relative prevalence of any given mutation compared with all mutant and wild-type sequencing reads, quantified as the allelic fraction (AF), a scale that most assays can use. AF calculations are highly correlated between NGS and ddPCR assays (Fig. 2),2 but it is unknown whether benign processes could spontaneously alter the calculation of AF. For example, it is possible that a change in the levels of wild-type DNA (for example, an acute infection leading to white blood cell degranulation) could result in fluctuations in mutation AF without any corresponding change in tumor burden.
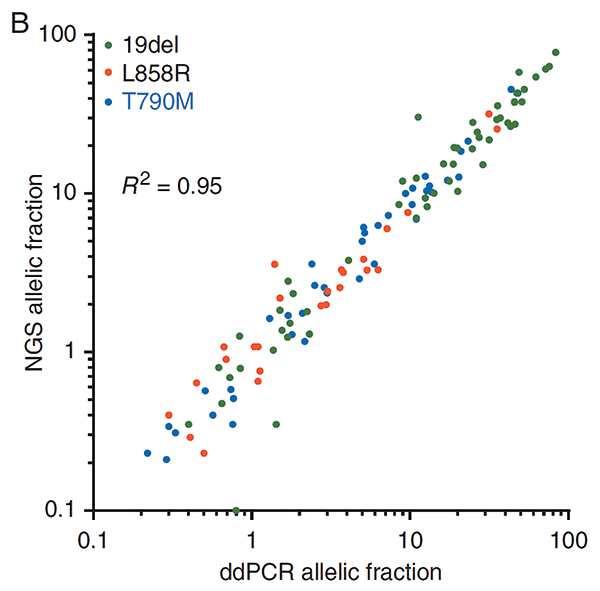
Abbreviations: ddPCR, droplet digital polymerase chain reaction; NGS, next-generation sequencing.
How to Track Multiple Variants
Earlier assays like ddPCR follow the single key driver mutation in plasma, whereas many newer assays use NGS, which can detect multiple mutations. Some of these mutations are cancer drivers, and some are subclonal mutations or resistance mutations. Furthermore, some mutations in cfDNA are germline; others are somatic mutations derived from clonal hematopoiesis and not from the tumor.3,4 How are these multiple variants best handled? Some investigators have tracked just the highest AF variant, although it may not be the driver mutation. Others have averaged the level of all variants at each timepoint. No consensus has been clearly established.
How Much Change Is Meaningful?
We know that complete clearance of mutations from plasma cfDNA is a good prognostic sign,5 but it is unclear what lesser magnitude of response is the best marker of treatment effect. There are decades of historical precedent supporting specific objective criteria for response and progression on tumor imaging.6 No such literature exists yet for response in plasma cfDNA. Some degree of change is likely due to random variation, unrelated to treatment effect. This must be robustly quantified so that clinicians can know what degree of change instead might be clinically meaningful. It would be unfortunate to change therapy based on variations in a blood test that are, in fact, due to assay artifact or extrinsic, nonmalignant conditions.
What Turnaround Time Is Needed?
Many have shown that plasma cfDNA analysis is much faster than getting a biopsy7; however, cfDNA testing remains slower than imaging or serum tumor markers. These tests involve multiple steps—spinning the plasma, extracting DNA, genomic analysis, and test interpretation—which can take days to weeks depending on the assay. This may be too slow for routine use because decisions about whether to continue treatment or to switch to a different regimen are usually made in a day or two based on imaging studies. Faster assays for cfDNA analysis are needed and are in development. In the meantime, turnaround time must be considered when building this testing into clinical decision making.
What Cost Will Be Scalable
Genomic analysis of plasma cfDNA can cost hundreds or thousands of dollars per specimen. This is likely too expensive for routine use every few months during therapy, unless clear clinical utility is established. For this reason, development of the cfDNA analysis as a monitoring assay should focus on key decision points where the cost–benefit ratio is more favorable. Of course, if cost decreases, it could be possible to run these tests more routinely.
Conclusions
Cancer monitoring using plasma cfDNA is compelling, but there is much work to be done. I currently do not use plasma cfDNA analysis on its own to assess response or progression, but at times I will use it in combination with imaging and clinical evaluation to help understand if a treatment is failing. Indeed, this also is how serum tumor markers are used—not on their own, but as a complement to imaging to understand the clinical picture. If scans seem mostly stable without response but symptoms are worsening, I might send plasma genotyping to assess for resistance. If I see high levels of the driver mutation, this is concerning because progression is likely brewing and makes me favor a change in treatment. However, if scans and symptoms are stable, a blood test on its own, especially one showing low AF mutations, is not enough to divert me from a treatment that is otherwise effective in the palliation of metastatic cancer. ✦
Acknowledgement: Dr. Oxnard is the Damon Runyon-Gordon Family Clinical Investigator supported by the Damon Runyon Cancer Research Foundation (CI-86-16).
About the Author: Dr. Oxnard is a physician with the Lowe Center for Thoracic Oncology Dana- Farber Cancer Institute and an associate professor of medicine at Harvard Medical School.
References:
1. Oxnard GR, Paweletz CP, Kuang Y, et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin Cancer Res. 2014;20(6):1698-1705.
2. Guibert N, Hu Y, Feeney N, et al. Ampliconbased next-generation sequencing of plasma cell-free DNA for detection of driver and resistance mutations in advanced non-small cell lung cancer. Ann Oncol. 2018;29(4):1049-1055.
3. Hu Y, Ulrich BC, Supplee J, et al. False-Positive Plasma Genotyping Due to Clonal Hematopoiesis. Clin Cancer Res. 2018;24(18):4437-4443.
4. Slavin TP, Banks KC, Chudova D, et al. Identification of Incidental Germline Mutations in Patients With Advanced Solid Tumors Who Underwent Cell-Free Circulating Tumor DNA Sequencing. J Clin Oncol. 2018 Oct 19. [Epub ahead of print].
5. Mok T, Wu YL, Lee JS, et al. Detection of EGFR-activating mutations from plasma DNA as a potent predictor of survival outcomes in FASTACT 2: A randomized phase III study on intercalated combination of erlotinib (E) and chemotherapy (C). J Clin Oncol. 2013;31(15)8021-8021.
6. Oxnard GR, Morris MJ, Hodi FS, et al. When Progressive Disease Does Not Mean Treatment Failure: Reconsidering the Criteria for Progression. J Natl Cancer Inst. 2012;104(20):1534-1541.
7. Sacher AG, Paweletz C, Dahlberg SE, et al. Prospective Validation of Rapid Plasma Genotyping for the Detection of EGFR and KRAS Mutations in Advanced Lung Cancer. JAMA Oncol. 2016;2(8):1014-1022.








{kind=link}