
Modified from Parslow AC et al. Biomedicines. 2015;4:4.
(A) Antibody-drug conjugate (ADC) accesses antigen via circulation; (B) ADC binds to antigen; (C) ADCantigen complex is internalized; (D) ADC-antigen complex is incorporated into endosomal vesicles; (E) ADC-antigen complex is processed along the endosomal-lysosomal pathway; (F) ADC is degraded in an acidic and proteolytic rich environment; (G) Cytotoxic payload is released intracellularly.
By David E. Gerber, MD
Posted: November 12, 2019

The underlying concept for antibody–drug conjugates (ADCs) is relatively straightforward: capitalize on the highly specific targeting of monoclonal antibodies to convey a lethal payload to cancer cells while minimizing exposure of normal tissues. However, after more than 20 years of clinical development, ADCs have not achieved their potential. Only recently have ADCs for NSCLC demonstrated promising effects in clinical trials, although none is yet approved for this malignancy.
Mechanism and Characteristics
To understand the challenges facing ADC development in NSCLC and other malignancies, it is worth reviewing their complex, multistep mechanism of action (Fig. 1). In an optimal scenario, ADCs extend the therapeutic window. Compared to conventional chemotherapy, they are designed to both increase efficacy and decrease toxicity. Specifically, targeted delivery of drugs to cancer cells results in increased drug doses in the tumor microenvironment, thereby lowering the minimum effective dose. Conversely, fewer drug molecules within normal, nontarget tissues lead to an increased maximum tolerated dose.1 Both tumor and ADC characteristics affect efficacy and toxicity. Ideal tumor characteristics include: high expression of the target antigen on the tumor surface, limited expression of the target antigen in healthy tissues, no shedding of the target antigen at high levels of expression, and a target antigen–ADC complex that is internalized upon binding. Desired ADC characteristics include: an antibody that has high affinity and avidity for the target antigen, a linking protein, and a payload (i.e., a highly potent drug) that is stable in circulation but is efficiently released inside of the tumor cells.2
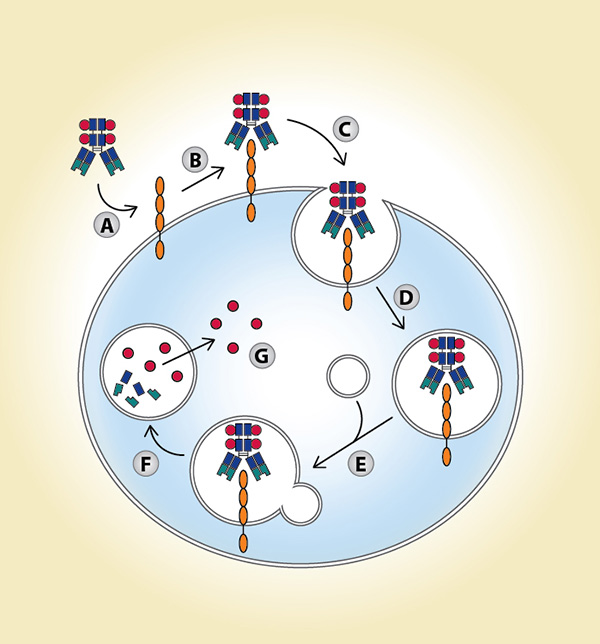
Modified from Parslow AC et al.
Biomedicines. 2015;4:4.
(A) Antibody-drug conjugate (ADC) accesses antigen via circulation; (B) ADC binds to antigen; (C) ADCantigen complex is internalized; (D) ADC-antigen complex is incorporated into endosomal vesicles; (E) ADC-antigen complex is processed along the endosomal-lysosomal pathway; (F) ADC is degraded in an acidic and proteolytic rich environment; (G) Cytotoxic payload is released intracellularly.
Design and Components
ADCs are composed of a monoclonal antibody, a linker, and a cytotoxic payload (drug; Fig. 2). Optimal linkers for ADC have a suitable site for attachment to the antibody, a complementary attachment site to achieve the rate of desired drug loading per antibody molecule, stability to process chemistry and product storage, and stability in circulation. They should also be cleavable after cellular internalization.3 ADC linkers may be cleaved through lysosomal proteases, which recognize and cleave a dipeptide bond to release free drug from conjugate, (e.g., valine-citrulline linker) or through pH-dependent hydrolysis, whereby acid labile groups within the linker are cleaved in the acidic environment of the lysosome (e.g., hydrazine linker). Alternatively, non-cleavable linkers remain attached to the ADC payload but depend on complete antibody degradation after ADC internalization. In general, non-cleavable linkers have greater stability in circulation and slower drug deconjugation than do cleavable linkers.2
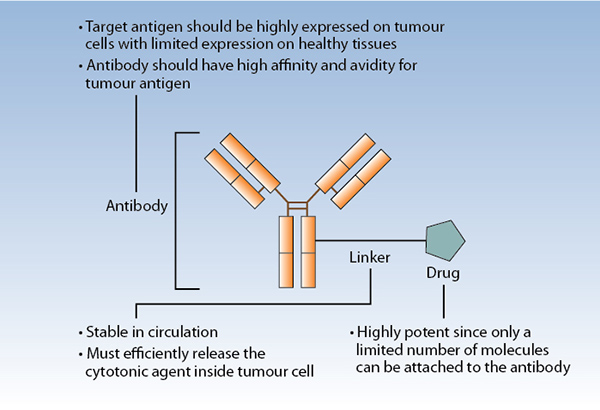
The optimal payload has high potency to prove efficacious at achievable intracellular concentrations, selectivity in killing target cells, low immunogenic potential, compatibility with the conjugation process, chemical stability in circulation, and either activity in linked form (for noncleavable linker systems) or in free form (for cleavable linker systems).3 Drugs employed as toxic payloads in ADCs generally fall into two categories: microtubule inhibitors (i.e., auristatins and maytansines) or DNA-damaging agents (i.e., calicheamicins, duocarmycins, pyrrolobenzodiazepine dimers, indolinone benzodiazepines, anthracyclines, and topoisomerase inhibitors). The majority of ADCs currently in development incorporate maytansine derivatives (DM1, DM4) or auristatins (MMAE, MMAF) as the chemotherapeutic component (Table). The desired drug–antibody (DAR) is generally 2:1 to 4:1, with higher ratios resulting in diminished stability and lower ratios resulting in reduced potency.
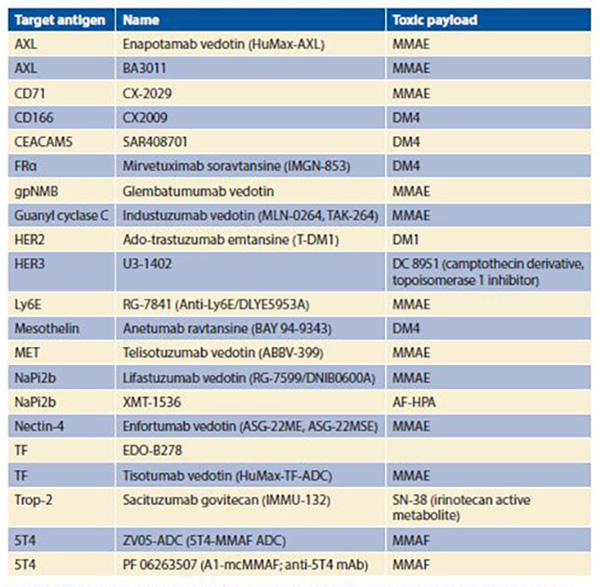
Abbreviations: AF-HPA, auristatin F-hydroxypropylamide; CEACAM5, carcinoembryonic antigen-related cell adhesion molecule 5; FRα, folate receptor α; HER2, human epidermal growth factor receptor 2 (ErbB2); HER3, human epidermal growth factor receptor 3 (ErbB3); Ly6E, lymphocyte antigen 6 complex, locus E; MMAE, monomethyl auristatin E; MMAF, monomethyl auristatin F; NaPi2b, sodium-dependent phosphate transport protein 2b; TF, tissue factor (thromboplastin; CD142); Trop-2, tumor-associated calcium signal transducer 2.
Resistance and Toxicity
Resistance to ADCs can arise from decreased antigen expression on the targeted cell surface due to either decreased target gene expression or increased mutations. Resistance can also be caused by decreased ADC internalization (due to decreased cell-surface trafficking or recycling), or multidrug resistance (MDR) transporter efflux out of the targeted cell. MDR transporter efflux can also exacerbate killing effects on bystander cells in the vicinity of the targeted cell, thereby potentially enhancing toxicity. Other potential reasons for ADC toxicities include target-dependent uptake and catabolism of the ADC, or release of free drug by deconjugation of circulating ADCs.
Toxicities from target-dependent ADC uptake occur when the target antigen is expressed in healthy tissues. Although not the dominant mechanism of toxicity, when related adverse events do occur, they may be profound. In some instances, previously unrecognized target-antigen expression on healthy tissues has led to major toxicities. Due to target-antigen expression on normal gastric mucosa, an ADC directed against the LewisY antigen resulted in hemorrhagic gastritis. Similarly, expression of CD446v6 in the deep layers of the skin led to fatal exfoliation from a CD446v6-directed ADC, and CA9 expression in intestinal mucosa resulted in fatal gastrointestinal toxicity from a CA9-directed ADC. Why might such surprises occur? Preclinical models may not adequately predict clinical activity and tolerability. Specifically, in some models the target antigen may not be expressed in host tissues, resulting in misleadingly favorable activity. Less severe but more common, antigen-independent toxicities reflect the inherent adverse effects of the payload, such as myelosuppression and neuropathy for microtubule inhibitors.
Clinical Development
As of March 2019, the U.S. Food and Drug Administration has approved three ADCs for the treatment of other malignancies: gemtuzumab ozogamicin (anti-CD33 antibody conjugated to calicheamicin) in acute myeloid leukemia, ado-trastuzumab emtansine in HER2-positive breast cancer, and brentuximab vedotin (anti-CD30 antibody conjugated to MMAE) in lymphoma. Adotrastuzumab provides an example of the therapeutic benefit of ADCs, as it has demonstrated superior efficacy compared to the unconjugated anti-HER2 antibody trastuzumab. Trials are now evaluating this agent in HER2-mutated NSCLC.
For the treatment of thoracic malignancies, perhaps the best-known ADC is rovalpituzumab tesirine (Rova-T; SC16LD6.5). Rova-T targets delta-like ligand 3, which is expressed in more than 80% of SCLC. Although Rova-T had demonstrated highly promising results in earlier single-arm trials,4 the phase III MERU trial was recently closed because it did not meet its primary endpoint of survival benefit for Rova-T compared with placebo.
The Table provides a listing of selected ADCs under development in NSCLC. The diversity of targeted antigens is readily apparent, as is the overwhelming use of maytansine derivatives or auristatins as toxic payloads. As is the case for other molecularly targeted therapies (e.g., small molecule inhibitors and unconjugated monoclonal antibodies), development of biomarkers for the identification of patients most likely to benefit represents a key consideration. Furthermore, with a distinct mechanism of action, ADCs may benefit a slightly different population than do other therapies with the same molecular target. For instance, small molecules targeting MET have clinical benefit largely limited to those NSCLC cases harboring MET exon 14 skipping mutations (< 5% of NSCLC cases). By contrast, the anti-MET ADC ABBV-399 has demonstrated responses in c-Met– positive NSCLC defined by immunohistochemical expression (up to > 50% of NSCLC cases depending on cutoff). For MET and other molecular targets, non- ADC therapies require the alteration to represent a true oncogenic driver for efficacy. However, because the primary mechanism of ADC killing is delivery of a cytotoxic payload rather than pathway interruption, target expression may suffice for efficacy. Although broader biomarker-defined subsets may expand the pool of eligible patients, efficacy may not reach that of targeted therapies for truly oncogene-addicted tumors. Conversely, it has been suggested that, even for ADCs, the presence of cellular signal dependence (e.g., HER2 mutations rather than HER2 expression) could enhance antitumor response, in this case through preferential ADC binding and internalization.5
Comparing ADC effects across tumor types provides insight into the importance of sensitivity to the cytotoxic payload. In HER2-positive (IHC 2+ or 3+) breast cancer, trastuzumab emtansine (T-DM1) had a response rate of 44% and a median progression-free survival (PFS) of 9.6 months.6 In NSCLC, when HER2 status is assessed by IHC, responses to T-DM1 were observed only in those with 3+ expression, where the response rate was 20% and median PFS was 2.7 months.7 Even in the more restricted— and potentially more sensitive—population of HER2-mutant NSCLC, median PFS was 5 months.5 Unfortunately, multiple ADCs have demonstrated similar effects in NSCLC clinical trials, with 17% response rate for the anti-Trop2 ADC sacituzumab govitecan,8 and a 19% response rate for the anti-MET ADC telisotuzumab vedotin in MET-positive cases.9 These relatively disappointing results could reflect the limits of cytotoxic therapies as NSCLC treatment, particularly after exposure to multiple prior lines of therapy, as is the case for most ADC clinical trial populations.
Future Directions
Newer-generation ADCs offer improved stability in circulation and favorable payload release kinetics intracellularly. They also benefit from improved characterization of the optimal patient population through biomarker development. A growing number of clinical trials are capitalizing on the favorable toxicity profiles of some ADCs and evaluating them in combination with other therapies. Another potential area of future growth is consideration of alternative payloads beyond cytotoxic drugs. To date, however, results with existing ADCs have been relatively disappointing, and no ADC has yet been approved in advanced lung cancer. ✦
About the Author: Dr. Gerber is Professor of Internal Medicine and Population & Data Sciences at the University of Texas Southwestern Medical Center. Within the Harold C. Simmons Comprehensive Cancer at UT Southwestern, he serves as Associate Director for Clinical Research and as Co-leader of the Experimental Therapeutics Program.
References:
1. Nasiri H, Valedkarimi Z, Aghebati-Maleki L, Majidi J. Antibody-drug conjugates: Promising and efficient tools for targeted cancer therapy. J Cell Physiol. 2018;233(9):6441-6457.
2. Thomas A, Teicher BA, Hassan R. Antibody-drug conjugates for cancer therapy. Lancet Oncol. 2016;17(6):e254-e262.
3. Singh SK, Luisi DL, Pak RH. Antibody-Drug Conjugates: Design, Formulation and Physicochemical Stability. Pharm Res. 2015;32(11):3541-3571.
4. Rudin CM, Pietanza MC, Bauer TM, et al. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: a first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol. 2017;18(1):42-51.
5. Li BT, Shen R, Buonocore D, et al. Ado-Trastuzumab Emtansine for Patients With HER2- Mutant Lung Cancers: Results From a Phase II Basket Trial. J Clin Oncol. 2018;36(24):2532-2537.
6. Verma S, Miles D, Gianni L, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367(25):1783-1791.
7. Peters S, Stahel R, Bubendorf L, et al. Trastuzumab Emtansine (T-DM1) in Patients with Previously Treated HER2-Overexpressing Metastatic Non-Small Cell Lung Cancer: Efficacy, Safety, and Biomarkers. Clin Cancer Res. 2019;25(1):64-72.
8. Heist RS, Guarino MJ, Masters G, et al. Therapy of Advanced Non-Small-Cell Lung Cancer With an SN-38-Anti-Trop-2 Drug Conjugate, Sacituzumab Govitecan. J Clin Oncol. 2017;35(24):2790-2797.
9. Goldman J. Phase I study of ABBV-399 (telisotuzumab vedotin) as monotherapy and in combination with erlotinib in NSCLC. J Thorac Oncol. 2017;12(11):S1805-S1806.






: More Stars Needed in the Constellation")

{kind=link}