
Biosimilars have proven themselves to the U.S. FDA, but clinicians are still wary.
By Leah K. Lawrence
Posted: June 2018
Healthcare providers are more than familiar with the use of brand name and generic drugs, but the passage of the Biologics Price Competition and Innovation (BPCI) Act of 2009 introduced a new player in the world of pharmaceuticals: the biosimilar.
In 2017, the U.S. Food and Drug Administration (FDA) approved the first biosimilar for cancer treatment, Mvasi™ (bevacizumab-awwb, Amgen Inc.). Mvasi is a biosimilar to Avastin® (bevacizumab, Genentech) and was approved for treatment of patients with certain colorectal cancers, nonsquamous NSCLC, glioblastoma, metastatic renal cell carcinoma, and cervical cancer. Although approved, Mvasi is not yet available for clinical use. “There will be tremendous pressure to consider using biosimilars soon,” said Corey J. Langer, MD, FACP, professor of medicine at The Hospital of The University of Pennsylvania and editor of the IASLC Lung Cancer News. “The presumption is that they are equivalent, but there is always a kernel of doubt.”
According to Dr. Langer, oncologists have been “burnt” in the past thinking that new compounds are as good as or better than reference products, only to have problems emerge post-approval. “With bevacizumab, I have nearly 20 years of experience working with the drug,” he said. “I have a comfort level with it, and personally, I can’t immediately export that comfort level to a biosimilar.”
What Is a Biosimilar?
A biosimilar is a biologic product that is highly similar to and has no clinically meaningful differences from an existing FDA-approved reference product.
The BPCI created an abbreviated licensure pathway for biosimilars to come to market. According to the FDA, this pathway was established as a way to provide more treatment options, increase access to lifesaving medications, and potentially lower healthcare costs through competition. The BPCI was designed similarly to the Drug Price Competition and Patent Term Restoration Act of 1984 (Hatch-Waxman Act), which encouraged generic competition for drugs. However, in contrast to generic drugs, which are small molecule compounds made through chemical means, biosimilars are generally large, complex molecules produced through biotechnology in a living system, such as a microorganism, plant cell, or animal cell.
“Unlike generic products, with biosimilars the manufacturer has to start with their own cell system and create their own process to make each biologic product unique,” said Richard Markus, MD, vice president, Global Development at Amgen. “The biosimilar is highly similar, but a unique product according to that manufacturer’s cell system and process.”
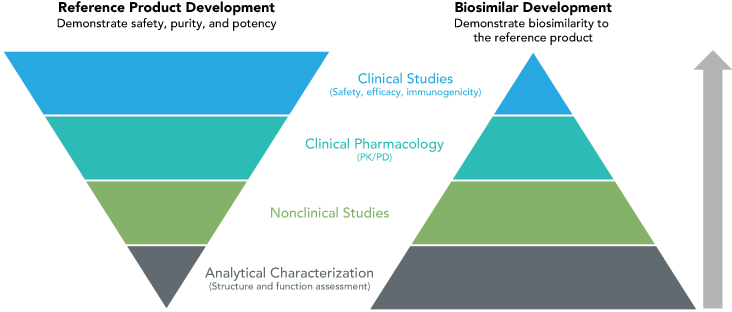
According to Dr. Markus, the FDA evaluates a manufacturer’s biosimilar product by looking at a totality of evidence that includes the analytic structure of the compound, functional and pharmacokinetic similarity, and clinical trial data that demonstrate equivalent efficacy, safety, and immunogenicity to the reference product (Fig.).
For Mvasi, Amgen conducted a phase III, double-blind clinical trial where patients with nonsquamous NSCLC were randomly assigned to Mvasi (328 patients) or Avastin (314 patients).1 The primary efficacy evaluation showed clinical equivalence between Mvasi and Avastin with a risk ratio of objective response rate for Mvasi compared with Avastin of 0.93 (90% CI [0.80, 1.09]) in the intent-to-treat population. In addition, the frequency, type, and severity of adverse events between the two biologics proved similar.
Based on the understanding of the mechanism of action of Mvasi, Amgen was able to obtain FDA approval for conditions other than nonsquamous NSCLC as well. Mvasi is approved as a biosimilar but not as an interchangeable product. An interchangeable biosimilar must meet additional requirements and, if approved, may be substituted for the reference product without involvement of the prescriber (Sidebar).
Clinicians’ Concerns
In addition to proven safety and efficacy, concern exists about the quality control of biologic biosimilars, according to Edgardo S. Santos, MD, FACP, medical director of cancer research and associate professor of clinical biomedical science at the Charles E. Schmidt College of Medicine at Florida Atlantic University.
“We don’t know if these products are produced in the United States or are coming from a global facility,” said Dr. Santos, who is also chair of the IASLC Publications Committee.
According to Dr. Markus, Amgen has manufacturing facilities in the United States but also in other locations throughout the world.
“All facilities have to meet FDA inspection as a biologic facility,” Dr. Markus said. “Amgen uses the same manufacturing network for biosimilars as it does for its [reference] products.”
In addition, although Mvasi is the only biosimilar for Avastin approved by the FDA, there are many more biosimilars of Avastin in development. That means that there may be choices available to the treating physician or hospital with respect to biosimilars.
Dr. Santos worries that a multitude of biosimilars for the same reference drug will cause confusion for clinicians.
“If a biosimilar is on the market, and clinicians start to use it and see similar efficacy to the reference product, I do not know why we must continue to invest in more biosimilars for the same compound,” Dr. Santos said. “We see the same phenomenon for reference drugs, and we end up with three or four drugs that all have the same efficacy for the same indication.”
However, additional options could help avoid a monopoly and promote competition for pricing, Dr. Santos added.
Cost
“Competition that can lower healthcare costs” is one of the intended outcomes of approving more biosimilars, according to a statement from FDA Commissioner Scott Gottlieb, MD, in the announcement of the approval of a second oncologic biosimilar Ogivri (trastuzumab-dkst, Mylan GmbH) for Herceptin® (trastuzumab, Genentech).2
A biosimilar company that is able to gain even a small share of the market for a biosimilar product will make a lot of money. Looking across all treatments, it is estimated that biologics can cost an average of more than $16,000 per year, a 20-fold increase from the $730 per year cost of traditional pharmaceuticals.3 At the midway point of 2016, Roche (which owns Genentech) reported a group revenue of approximately $25 billion supported largely in part by sales of three biologics: rituximab, trastuzumab, and bevacizumab.4
“Lowering costs is a laudable goal,” Dr. Langer said. “Oncology care is very expensive, potentially unsustainable, one could argue. Anything that can reduce the cost without sacrificing therapeutic efficacy would be welcome.”
However, Drs. Langer and Santos are both concerned that if biosimilars are introduced to the market at significant cost savings, then insurance payers may decide to no longer cover the higher cost of the reference product before the biosimilars have proven themselves in a real world setting.
According to Dr. Santos, this situation is already occurring for Neupogen® (filgrastim), a growth factor used in the treatment of neutropenia. The first biosimilar approved by the FDA was Zarxio (filgrastim-sndz), which is a biosimilar for Neupogen.
“There are insurance companies that do not allow use of Neupogen as growth factor support and not even Neulasta® (the pegylated form of filgrastim), which has shown better efficacy than Neupogen in head-to-head comparison with greater patient convenience,” Dr. Santos said. “This will occur for other drugs as well.”
According to Dr. Markus, Amgen has not announced a cost for Mvasi. However, he added that as oncologists and physicians begin to embrace biosimilars, they should not do it based on cost alone, and they should not do it blindly.
“Even though a product is approved, physicians will need to understand [biosimilars] a bit to feel confident about using them,” Dr. Markus said. “There may eventually be a choice in which commercially available biosimilar agent they use and they, or an expert in their hospital system, should make their choice based on evaluation of the quality of the data, the clinical trial designs, and the results. They should also consider the fact that these agents are manufactured in a quality facility, just like they would before using a new biologic agent.” ✦
Related Article: FDA Fosters Understanding about Approval Process for Biosimilars
References:
1. Thatcher N, et al. Randomized, double-blind, phase 3 study evaluating efficacy and safety of ABP 215 compared with bevacizumab in patients with non-squamous non-small-cell lung cancer. J Clin Oncol. 2016: 34(suppl 15):9095-9095.
2. FDA approves first biosimilar for the treatment of certain breast and stomach cancers. U.S. Food and Drug Administration website. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm587378.htm. Accessed February 15, 2018.
3. Shapiro RJ, Singh K, Mukim M. The Potential American Market for Generic Biological Treatments and the Associated Cost Savings. Sonecon LLC: Washington, DC, February 2008.
4. Pagliarulo N. Roche sales buoyed by cancer drugs. BioPharmaDive website. http://biopharmadive.com/news/roche-sales-buoyed-by-cancerdrugs/423032/. Accessed February 15, 2018.








{kind=link}